Ph.D. Materials Science and Engineering and Scientific Computing at University of Michigan; B.S. Physics at Idaho State University
I am a postdoc working at MIT with Prof. Tess Smidt and Prof. Jeffery Grossman to build better representations of quantum materials data. This increases our chemical understanding and improves machine learning architectures for simplified training.
My scientific interests center around decoding complicated quantum simulations into chemically intuition frameworks to reveal the covalent, ionic, and metallic bonds which conspire to shape in material properties.
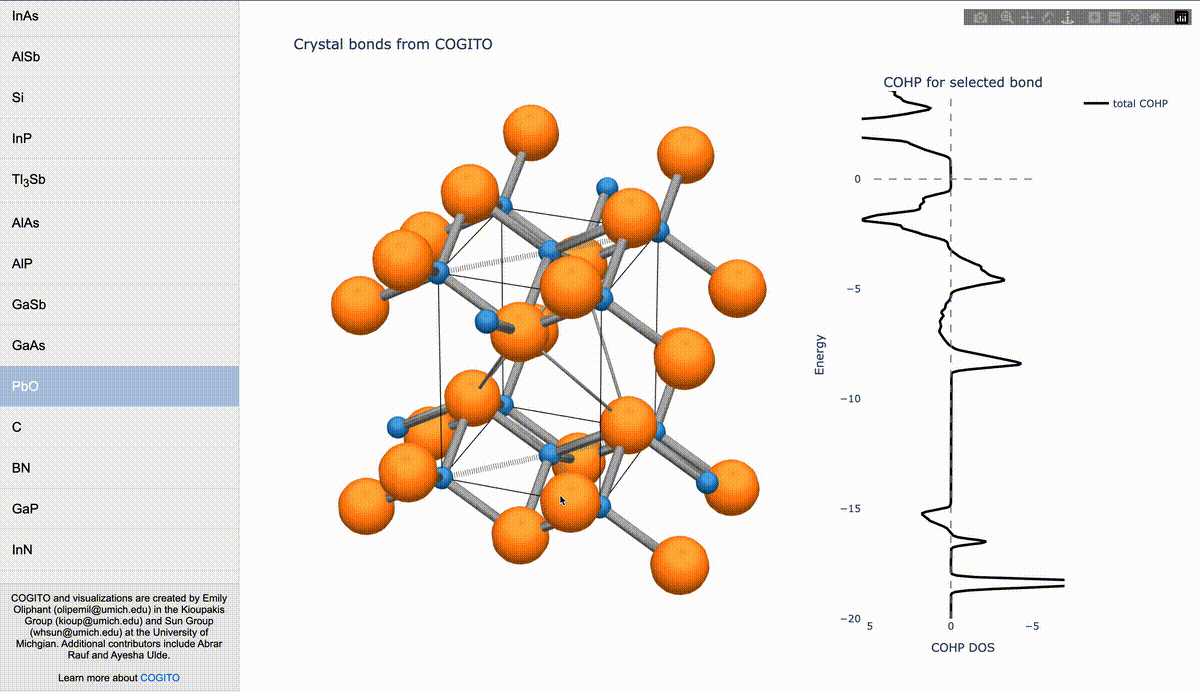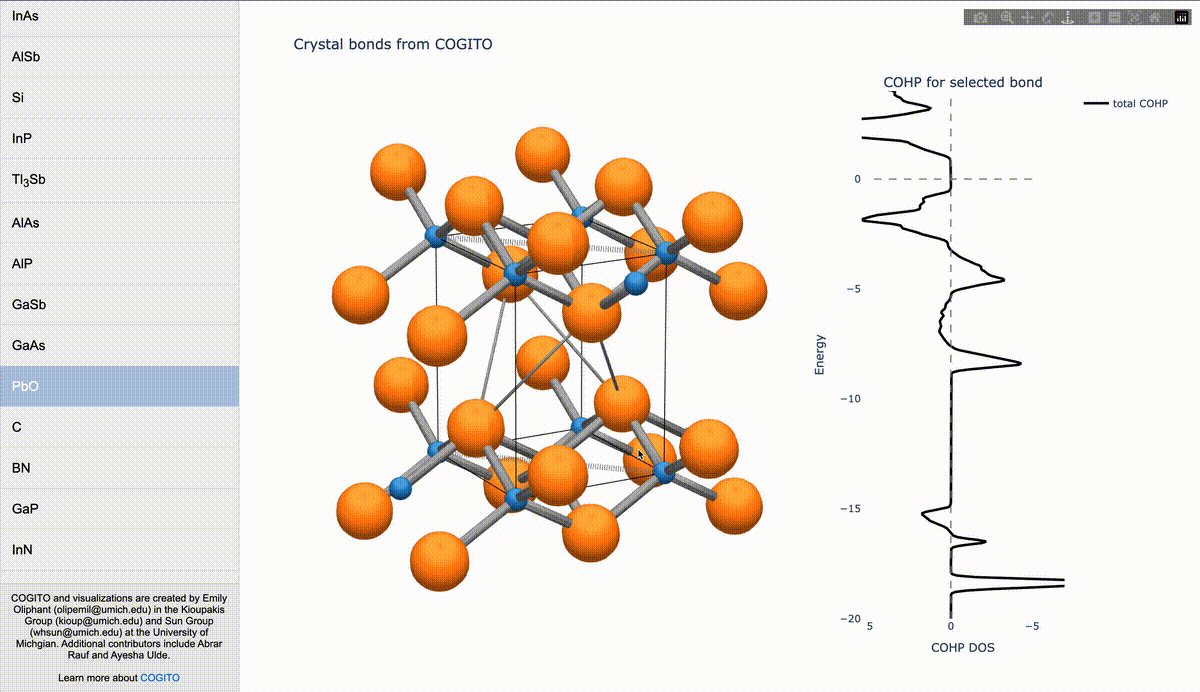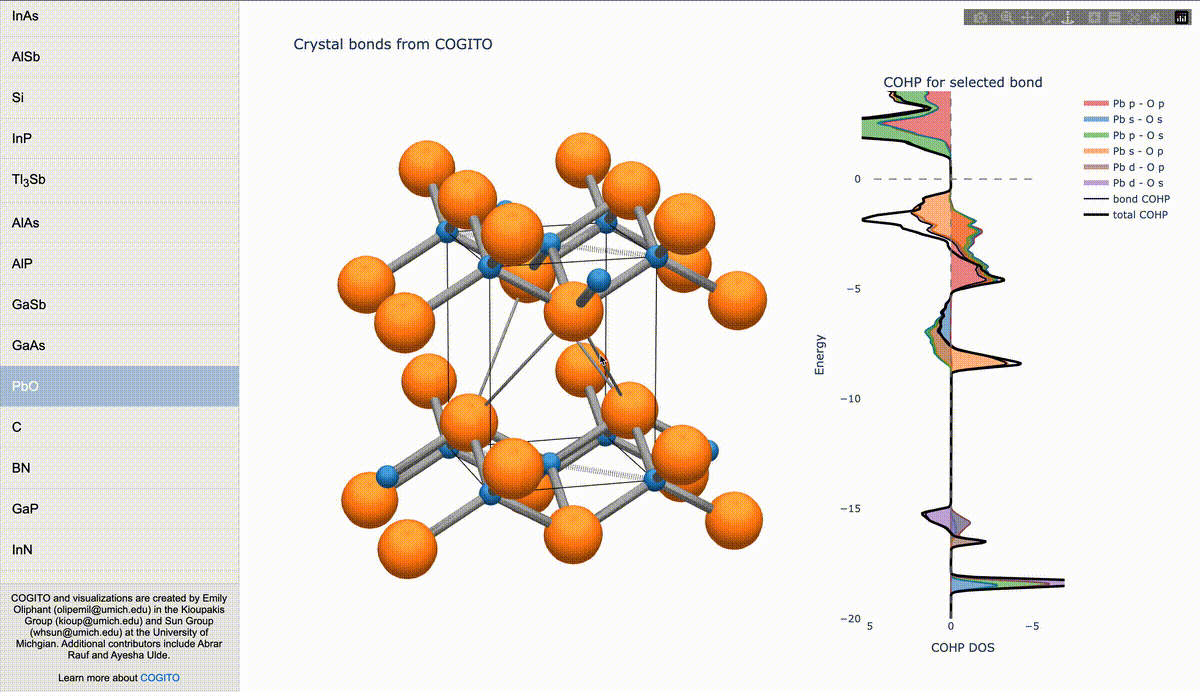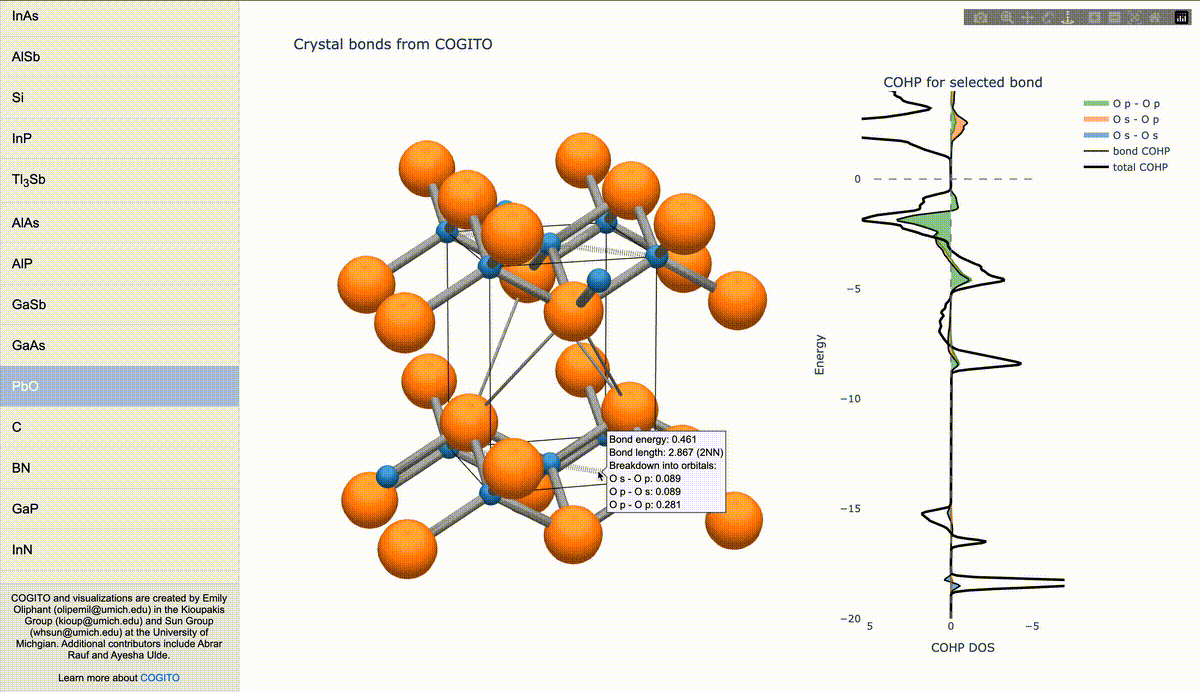
During my PhD, I created the toolkit Crystal Orbital Guided Iteration To atomic-Orbitals (COGITO). This combined the devleopment of novel algorithms to parse DFT wavefunctions into an adaptable, maximally unique atomic basis with a powerful user inferface to visualization bonding and charge transfer in crystals. For more information, check out the COGITO website or the COGITO paper. COGITO was created in the Sun group and Kioupakis group at University of Michigan.
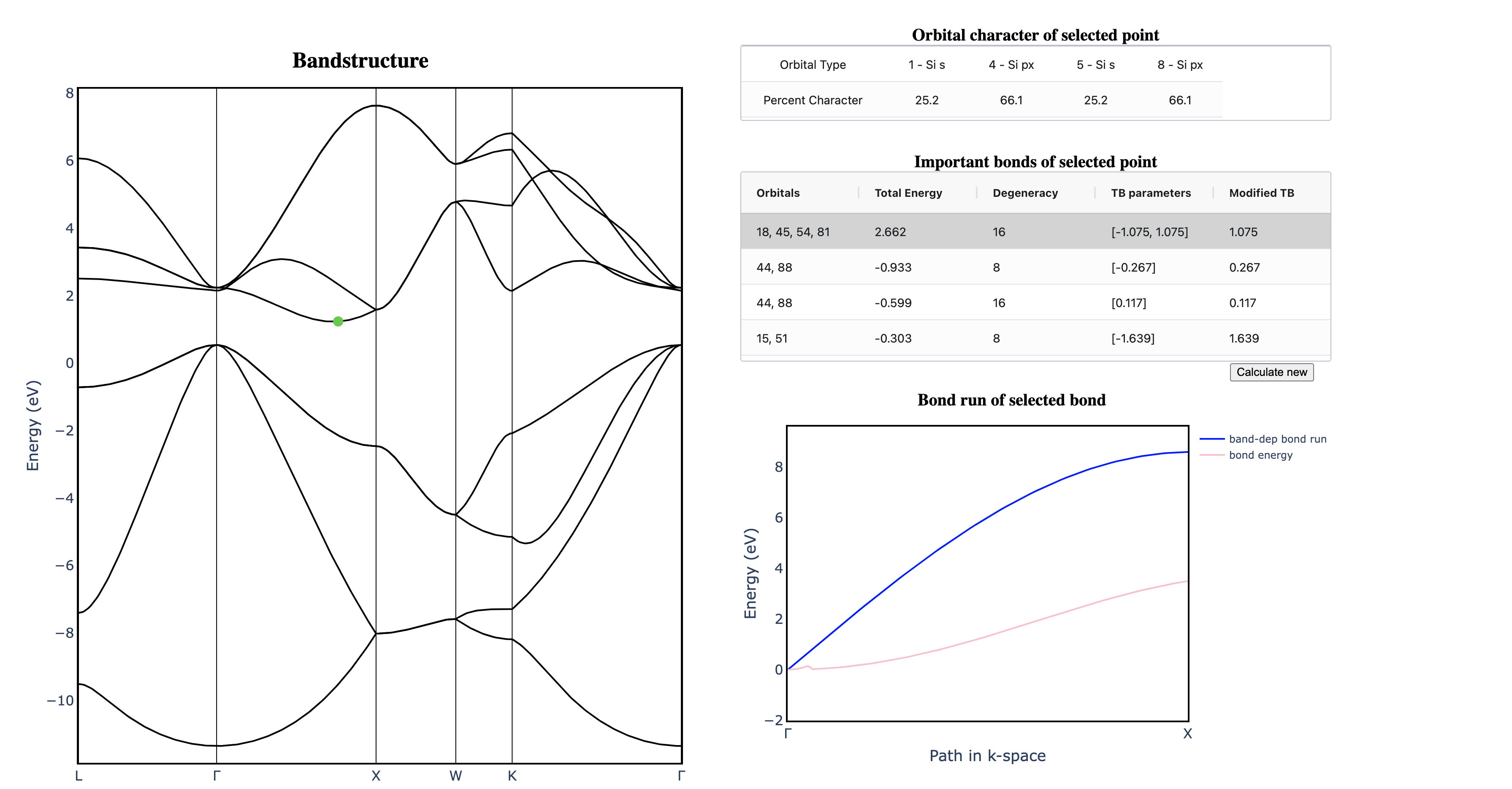
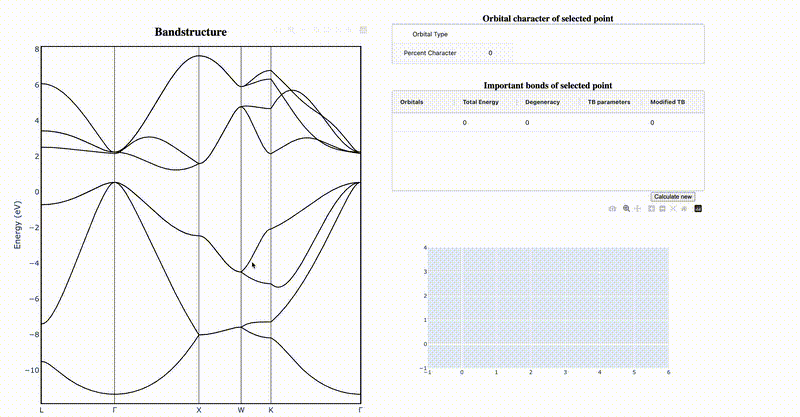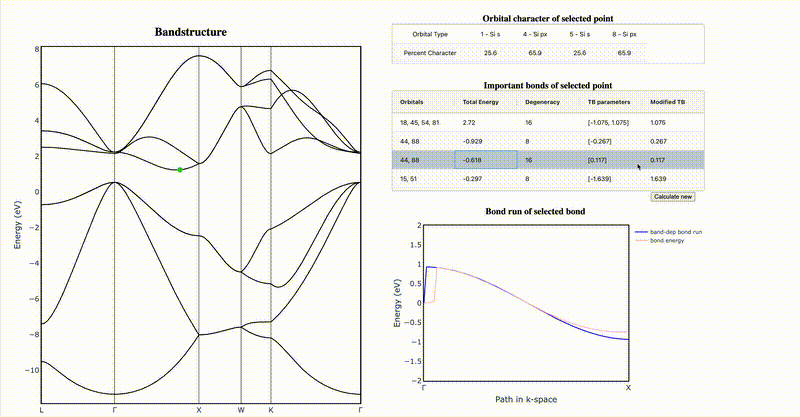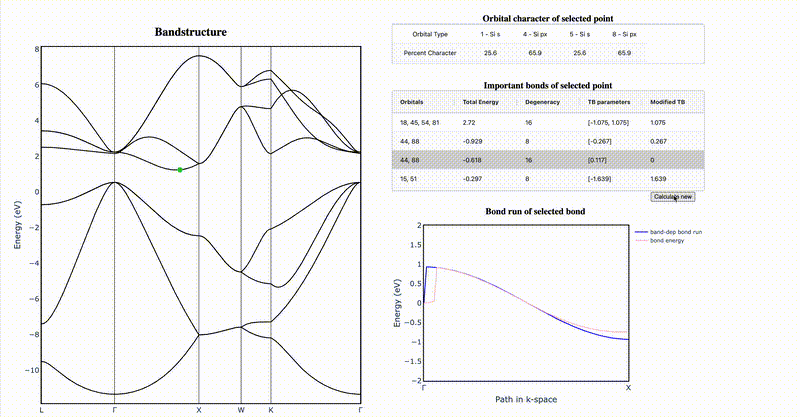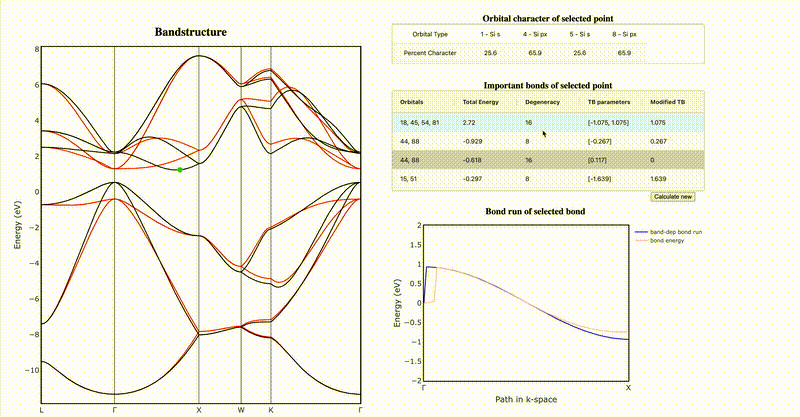
It is difficult to intuit how electronic structure features—such as band gap magnitude, location of band extrema, effective masses, etc.—arise from the underlying crystal chemistry of a material. Here we present a strategy to distill sparse and chemically-interpretable tight-binding models from density functional theory calculations, enabling us to interpret how multiple orbital interactions in a 3D crystal conspire to shape the overall band structure. Applying this process to silicon, we show that its indirect gap arises from a competition between first and second nearest-neighbor bonds—where second nearest-neighbor interactions pull the conduction band down from Γ to X in a cosine shape, but the first nearest-neighbor bonds push the band up near X, resulting in the characteristic dip of the silicon conduction band. By identifying the essential orbital interactions that shape the conduction band, we can further rationally tune bond strengths to morph the silicon band structure into the germanium band structure. Our computational approach serves as a general framework to extract the crystal chemistry origins of electronic structure features from density functional theory calculations, enabling a new paradigm of bonding-by-design.
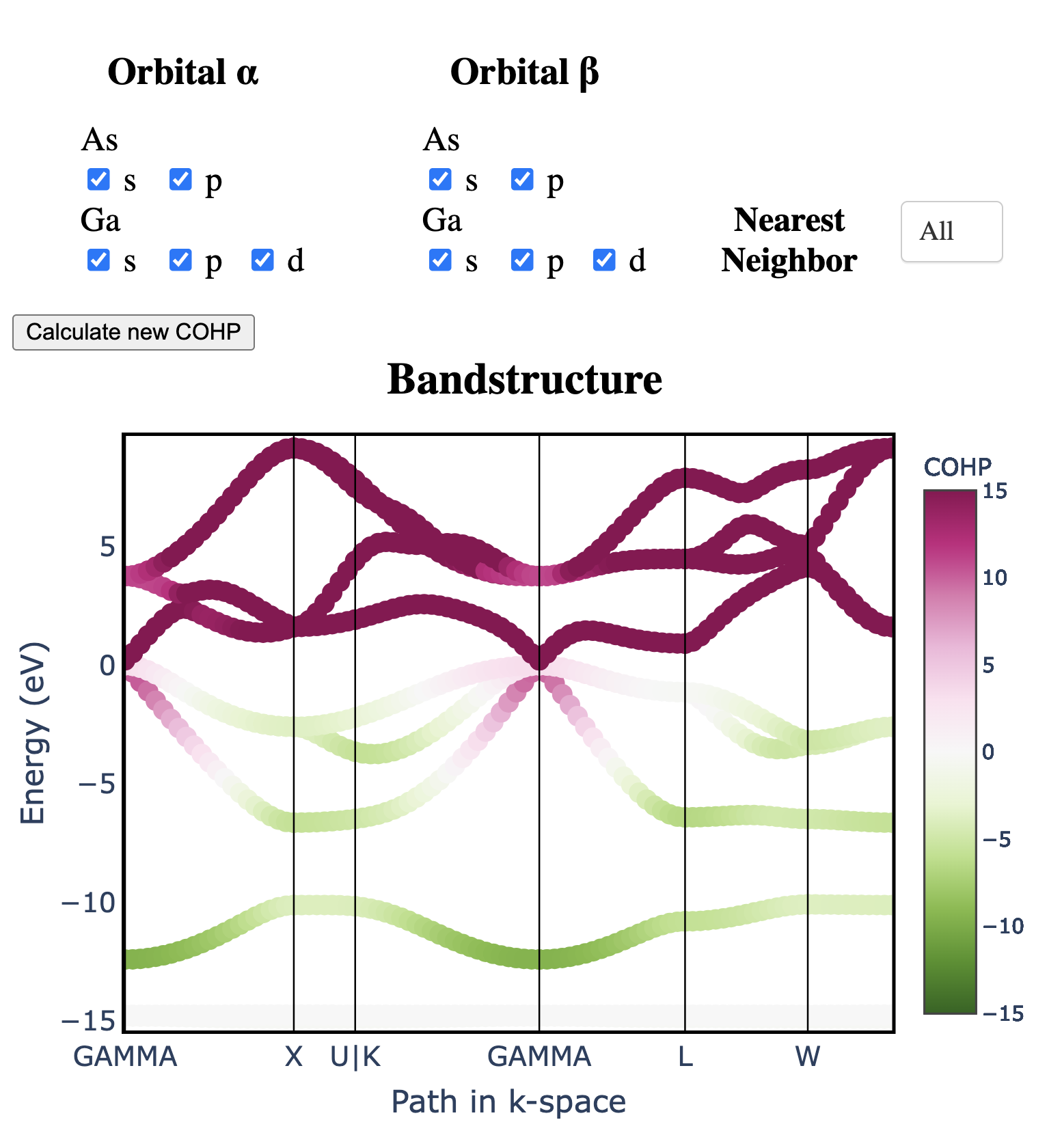
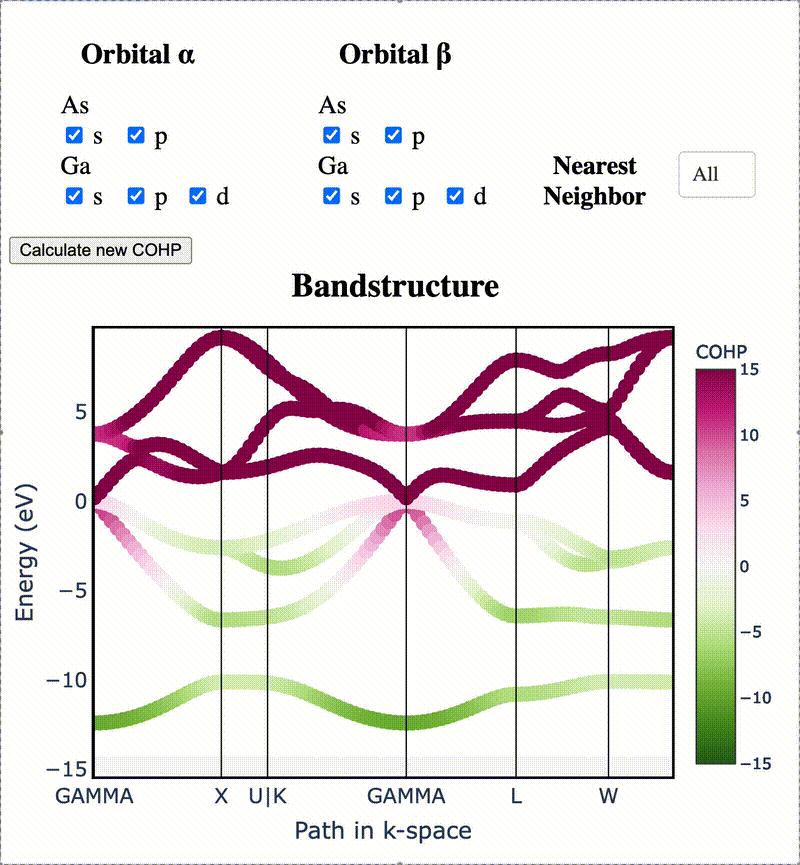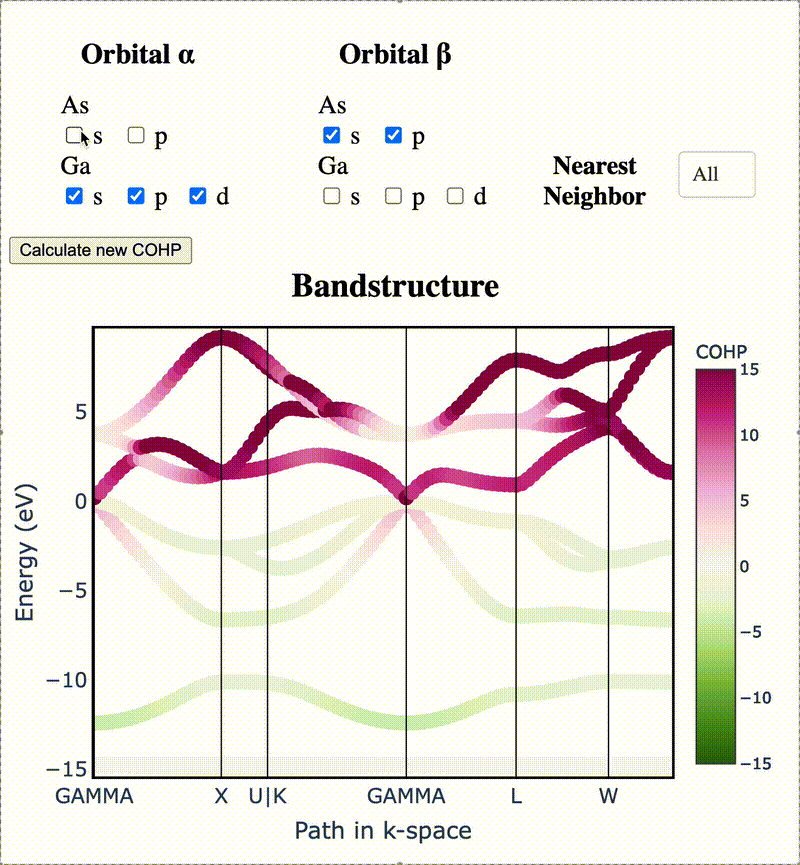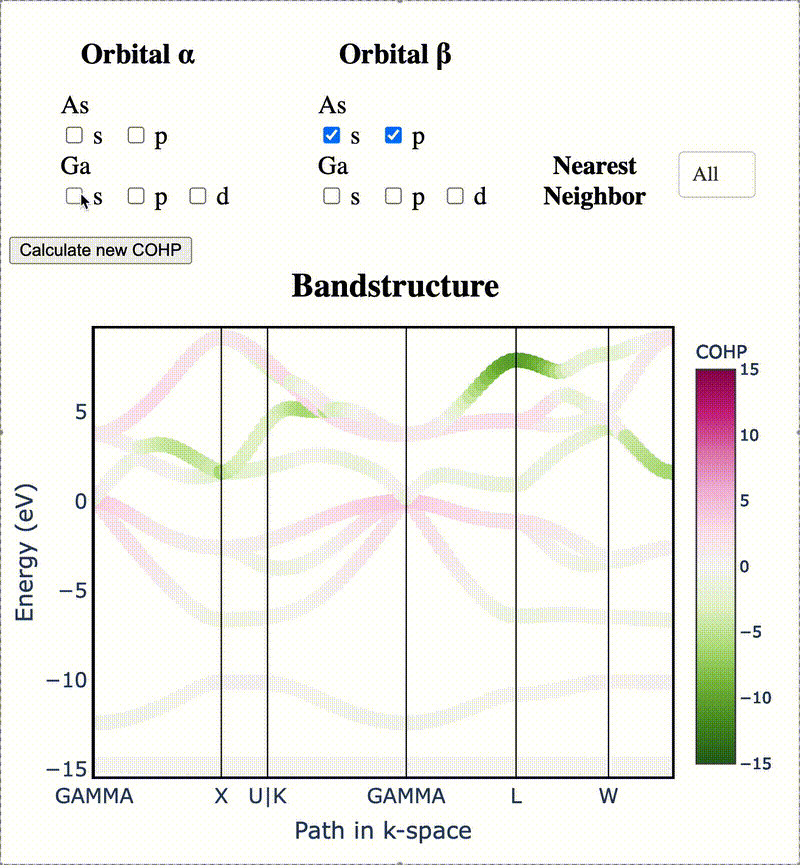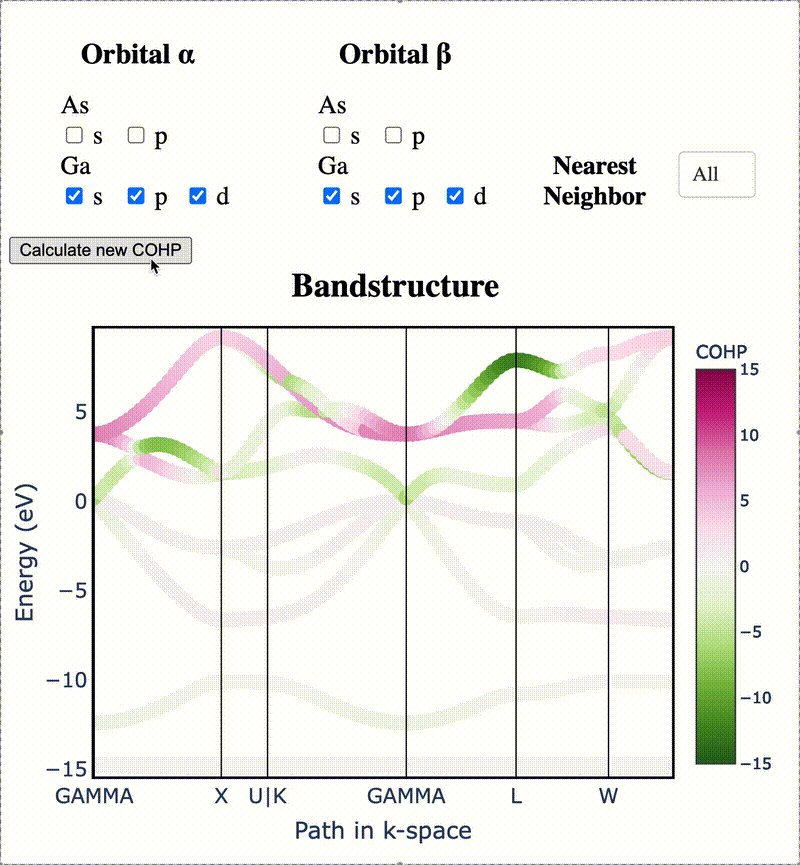
Atomic orbitals underpin our understanding of electronic structure, providing intuitive descriptions of bonding, charge transfer, magnetism, and correlation effects. Despite their utility, an atomic basis that is adaptable, strictly localized on atomic centers, and enables accurate tight-binding interpolation has remained elusive. Here, we introduce Crystal Orbital Guided Iteration To atomic-Orbitals (COGITO), a framework that constructs an optimal atomic orbital basis by identifying and resolving key mathematical obstacles inherent to nonorthogonal bases–particularly uncontrolled orbital mixing, and the fixed-overlap constraint between orbitals. We demonstrate that COGITO enables tight-binding models as accurate as MLWF-based approaches, while preserving the ability of tight-binding parameters to represent the projected atomic basis–an essential feature lost in schemes that enforce orbital orthogonality or maximal localization. By creating accurate and chemically interpretable models of electronic structure, COGITO reveals the orbital-resolved covalent bonds and charge transfer that is encoded in the Kohn-Sham wavefunctions of DFT. Our method thus offers a powerful tool for any physics- or chemistry-based application that relies on a faithful description of local electronic structure.
One of my passions is to create interactive visualizations that explore the connection between bonding and quantum mechanics. Please enjoy the visualizations below by clicking Launch Interactive for the interactive version and View Explanation for a beginner friendly background to what is displayed.

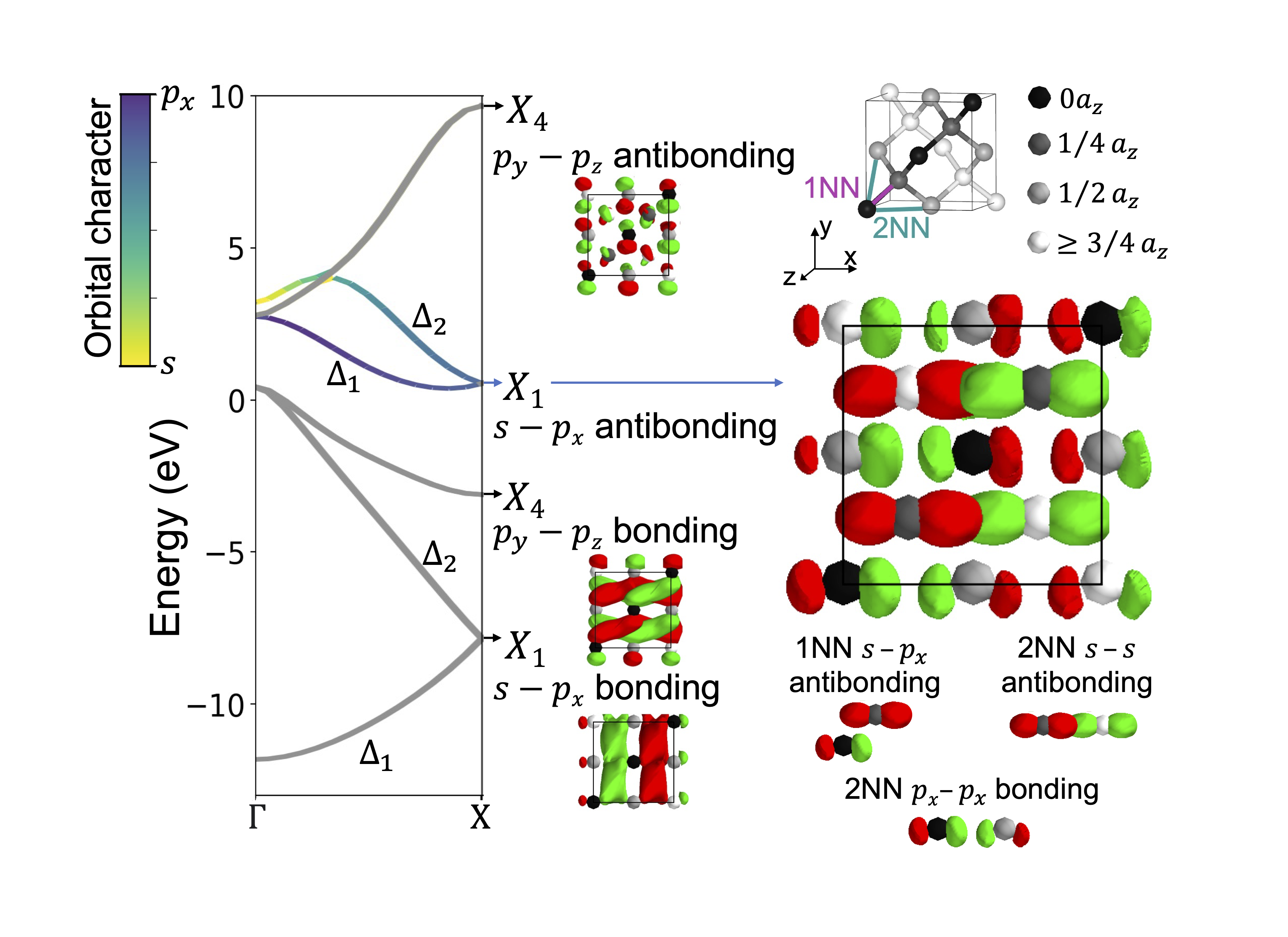 Why does silicon have an indirect band gap?
Why does silicon have an indirect band gap?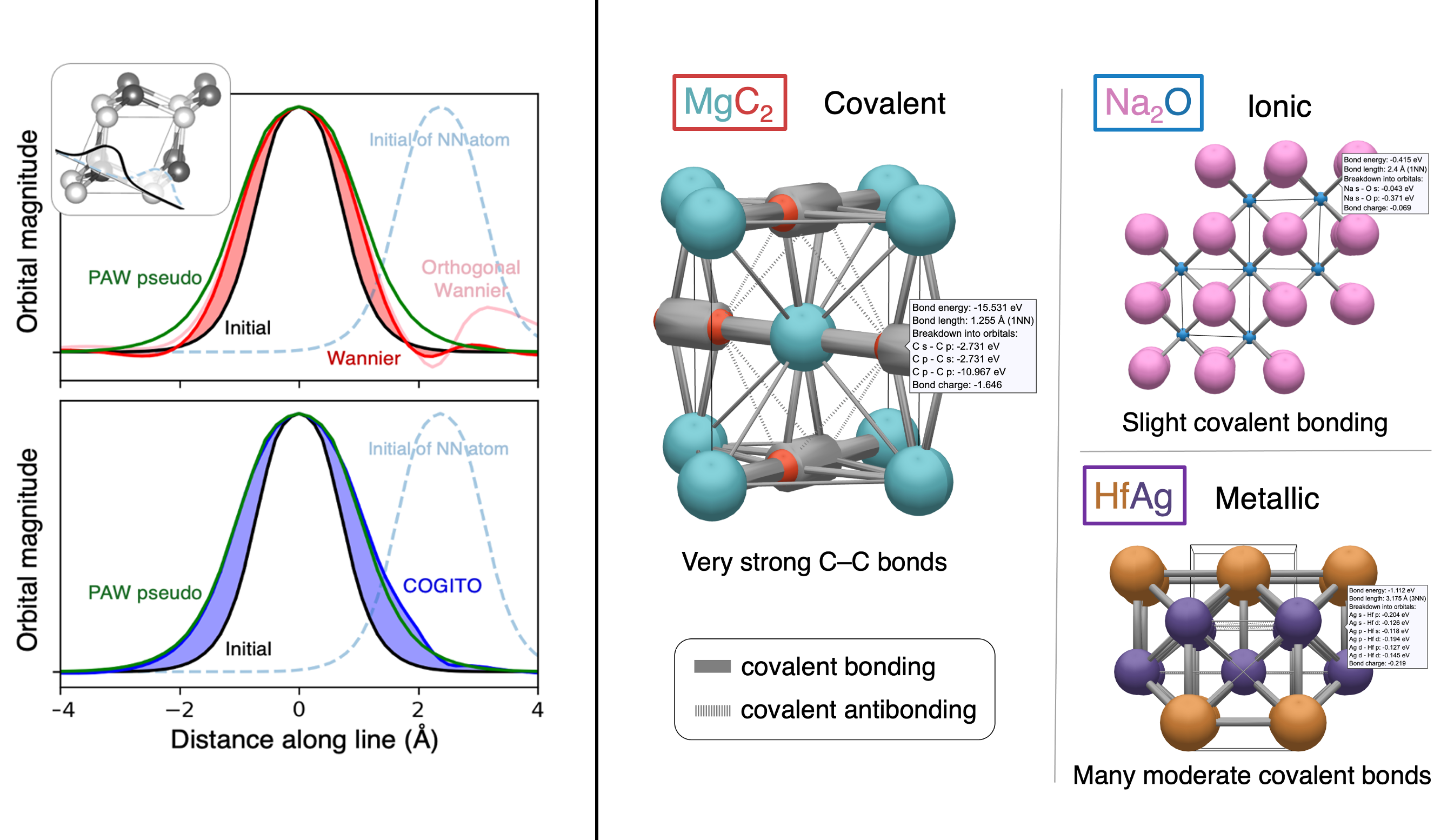 Crystal Orbital Guided Iteration to Atomic Orbitals
Crystal Orbital Guided Iteration to Atomic Orbitals